
13 Oct E. coli – From Human Stool to Biotech Tool – Part 2
In our previous installment of this series on E. coli, we explored the serendipitous discovery and adoption of this microbe as a model organism by the research community. In this installment, we will fast-forward to a more recent series of events that cemented E. coli’s place in the biotech industry.
The year was 1973, and a lot of important events where transpiring: the United States officially ended its involvement in the Vietnam War; Pink Floyd released their “Dark Side of the Moon” album, which would go on to become one of the best-selling musical albums of all time; and the first cellular telephone call was made. Few people are aware that this year also gave rise to the birth of “recombinant DNA technology” – the technological engine of the biotech industry. In November of 1973, several independent discoveries were combined in a creative way and published in a seminal scientific paper by Stanley Cohen, Annie Chang, Herbert Boyer, and Robert Helling titled, “Construction of biologically functional bacterial plasmids in vitro”1. Before diving into the main achievement of that paper, let’s lay the groundwork:
Plasmids
As early as the 1940s, scientists observed autonomous extrachromosomal DNA elements that were replicating inside certain bacteria. In 1952 Joshua Lederberg proposed the term “plasmid” to describe these extrachromosomal elements2. It wasn’t clear at the time what these plasmids were doing, but they were clearly able to replicate in the host and be inherited by daughter cells. Unlike bacterial chromosomes, which were very large (typically several million base pairs of DNA), plasmids could be quite small (some as small as just a few thousand base pairs), though we now know of the existence of plasmids that are on par with chromosomes in terms of size. At a minimum, a plasmid contains a special region of DNA that interacts with DNA replication proteins – a region commonly referred to as the “origin of replication”. Sometimes the plasmid carries the genes for its own replication proteins, while in other cases it relies on the native DNA replication machinery of the host. Additionally, plasmids usually carry genes that provide some functional benefit to their host, like genes that provide resistance to environmental stressors (such as antibiotics, heavy metals, or radiation), genes that endow the host with new metabolic capabilities (such as the ability to use new compounds as energy sources), genes that produce toxins that can kill other bacteria or predators in the local environment (thus reducing competition for the plasmid-carrying strains), or genes that allow the bacteria to colonize new environmental niches. Plasmids often also encode complex molecular machinery to facilitate their spread between hosts in the environment, ensuring their long-term propagation.
"Competent" Cells
Eventually, a method to introduce foreign DNA into E. coli was developed3. In this process, strains of E. coli that were treated with a salt solution that rendered them “competent” (i.e., able to take up DNA from their environment). When these competent cells were exposed to isolated bacteriophage DNA, the DNA was able to enter the cells where it began to function. This technique was eventually extended to plasmid DNA by Leslie Hsu and Annie Chang, enabling isolated plasmids to be introduced into new strains where they would take up residence and start replicating4.
DNA-Modifying Enzymes
In the 1960s and 1970s, two extremely important enzymes were isolated and characterized. First, in 1967, an enzyme named DNA ligase was isolated5. This enzyme, which plays a role in DNA replication and repair processes inside the cell, can take two separate fragments of DNA and chemically join them together into a single fragment. Next, in 1970 Hamilton Smith published his work on isolating and purifying a restriction endonuclease enzyme from the bacterial strain Haemophilus influenzae6–7. Restriction enzymes are so called because they were observed to “restrict” foreign DNA from functioning inside a host. Smith was able to demonstrate that these enzymes worked by recognizing a specific short sequence of DNA. If this specific short sequence of DNA was present in a longer stretch of double-stranded DNA, the restriction enzyme would bind to it, and acting like a pair of molecular scissors, cut through both strands of the DNA. These enzymes function as a kind of primitive immune system, cutting up the DNA of invading viruses and preventing infection. Following Smith’s work, a plethora of restriction enzymes were isolated from a myriad of bacterial species, each enzyme recognizing and cutting at a unique “restriction site” that is specific for that enzyme. For example, the restriction enzyme known as “EcoRI” recognizes the DNA sequence “GAATTC”.
Putting together all of the pieces
With this background information in place, we can return to November of 1973. This was the year that Cohen, Chang, Boyer, and Helling put all of these pieces together to create recombinant DNA technology.
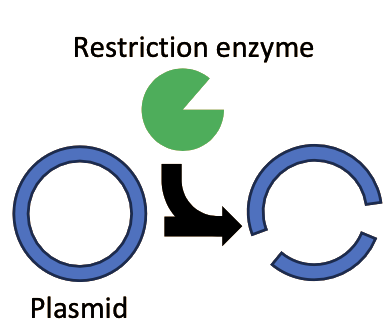
Step 1
An isolated plasmid encoding an antibiotic resistance gene could be treated with a restriction enzyme to cut the plasmid into multiple fragments. The specific DNA fragment containing the plasmids origin of replication and the antibiotic resistance gene could be isolated.

Step 2
A second, different plasmid could also be treated with a restriction enzyme, and a fragment from this plasmid could be isolated.
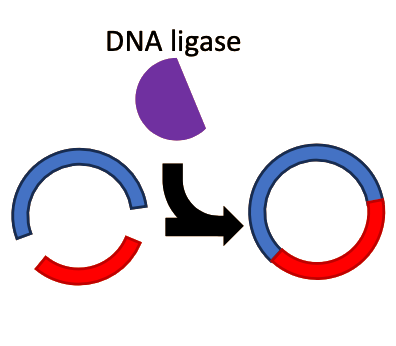
Step 3
The isolated fragments from these two separate plasmids could be mixed together and treated with DNA ligase. The ligase enzyme would “glue” these two fragments together into a new, chimeric plasmid.
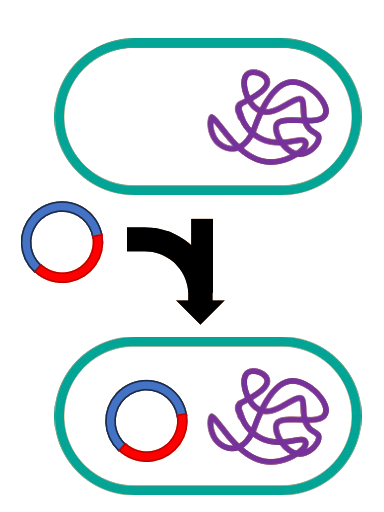
Step 4
The new Frankenstein plasmid could be introduced into E. coli cells that had been made competent, where the new plasmid would take up residence and begin replicating. Cells that had taken up this plasmid could be selected by growing the cells in the presence of the antibiotic that the plasmid encodes resistance to. Cells with the plasmid would be resistant to the antibiotic, while cells that failed to take up the plasmid DNA would be killed by the antibiotic.
This is exactly what this clever group of scientists was able to demonstrate in their 1973 paper. Subsequent papers showed that this technique could be used to “clone” genes from eukaryotic cells (genes from the frog Xenopus laevis) into plasmids, and that when introduced into E. coli, these foreign genes could be functionally transcribed inside the host.
The commercial applications of this line of research did not go unnoticed. Eventually, a venture capitalist named Robert Swanson was able to convince Herbert Boyer (one of the scientists involved in the work described above) to join him in the founding of a new biotechnology company called Genentech in 1976.
After first demonstrating in 1977 that the approach could be used to introduce a plasmid containing a synthetic gene for somatostatin into E. coli, leading to the production of this hormone9, the team set their sights on a loftier (and more commercially viable) use of the technology – to provide a reliable source of human insulin for diabetic patients. At the time, insulin for diabetics was sourced from cattle and pigs, where the pancreases from 23,500 animals were required to make one pound of insulin (to meet annual demand in the US for the drug, 56 million animals were required)10. Not only was the sustainability of sourcing insulin from animals questionable, but many patients developed allergies to the animal- sourced product.
Following a true tour de force (wonderfully chronicled in a book by Sally Smith Hughes11), the scrappy biotech startup published the successful production of human insulin in E. coli using recombinant DNA technology in 197912. The technology was subsequently licensed to Eli Lilly, which succeeded in getting recombinant human insulin approved by the FDA in 1982 (where E. coli-produced insulin is still the gold standard).
Since that time, recombinant DNA technology has seen major advances and has been applied in the pharmaceutical, diagnostic, energy, agriculture, and industrial chemicals industries. Thanks to new tools for DNA synthesis, what took the Genentech team months of painstaking, around-the-clock effort has effectively become commoditized, now being completed by numerous commercial vendors in a few days for a few hundred dollars. Using DNA sequencing technologies an entire human genome can now be sequenced in just a few hours for hundreds of dollars (compared to the multi-billion-dollar effort to sequence the first human genome). However, when it comes to cloning hosts (the organisms that plasmid DNA is constructed and propagated in), scientists are still using the same E. coli K-12-derived cloning hosts that were used in the late 1970s.
Part of this lack of innovation might stem from an “if it ain’t broke, don’t fix it” mentality. While it is true that traditional cloning hosts have served the biotech industry well for many decades (where the primary need was to clone simple genetic constructs), there has been an increase in demand for plasmids carrying larger, more complex genetic payloads. These plasmids are pushing existing cloning hosts to their limits (resulting in costly manufacturing challenges) and creating an opportunity for innovation. In our next white paper, we will explore the numerous challenges faced in modern plasmid DNA production.
References
- Cohen, S.N.; Chang, A.C.Y.; Boyer, H.W.; Helling, R.B. 1973. Construction of biologically functional bacterial plasmids in vitro. Proc. Natl. Acad. Sci. USA. 70(11):3240–3244
- Lederberg, J. 1952. Cell genetics and hereditary symbiosis. Physiol. Rev. 32(4):403–430
- Mandel, M.; Higa, A. 1970. Calcium-dependent bacteriophage DNA infection. Journal of Molecular Biology. 53(1):159–162
- Cohen S.N.; Chang A.C.Y.; Hsu, L. 1972. Nonchromosomal antibiotic resistance in bacteria: Genetic transformation of Escherichia coli by R-factor DNA. Proc. Natl. Acad. Sci. USA. 69(8):2110–2114
- Shuman, S. 2009. DNA Ligases: Progress and prospects. J. Biol. Chem. 284(26):17365–17369
- Smith, H.O.; Wilcox, K.W. 1970. A restriction enzyme from Hemophilus influenzae. I. Purification and general properties. J. Mol. Biol. 51:379–391
- Kelly, T.J., Jr.; Smith, H.O. 1970. A restriction enzyme from Hemophilus influenzae. II. J. Mol. Biol. 51:393–409
- Morrow, J.F.; et al. 1974 Replication and transcription of eukaryotic DNA in Escherichia coli. Proc. Natl. Acad. Sci. USA. 71(5):1743–1747
- Itakura, K.; Hirose, T.; Crea, R.; Riggs, A.D.; Heyneker, H.L.; Bolivar, F.; Boyer, H.W. 1977. Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin. Science. 198(4321):1056–1063
- https://www.gene.com/stories/cloning-insulin
- Hughes, S.S.; 2013. Genentech: The Beginnings of Biotech. University of Chicago Press; Reprint edition. ISBN-13: 978-0226045511
- Goeddel, D.V.; Kleid, D.G.; Bolivar, F.; Heyneker, H.L.; Yansura, D.G.; Crea, R.; Hirose, T.; Kraszewski, A.; Itakura, K.; Riggs, A.D. 1979. Expression in Escherichia coli of chemically synthesized genes for human insulin. Proc. Natl. Acad. Sci. USA. 76(10):106–110